Protein misfolding is at the root of many neurodegenerative diseases of ageing, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Creutzfeldt-Jakob disease (CJD). These are diseases of protein conformation: the misfolding and subsequent aggregation of proteins or peptides into β-sheet-enriched forms initiates a cascade of events that ultimately leads to neurological dysfunction and death of neurons or glial cells in the brain. There is not a single therapeutic that is effective at halting or even slowing the progression of any of these diseases. Thus, understanding the earliest disease-associated events (i.e. the initial formation of misfolded proteins) is paramount to being able to develop an effective treatment.
My lab focuses on understanding the molecular details of protein aggregation with an emphasis on designing accurate animal models of protein misfolding diseases.
Interrogating the Mechanisms of Spontaneous Prion Formation and Prion Neurotoxicity
Prions are infectious proteins that cause a variety of rare but invariably fatal neurodegenerative illnesses such as CJD in humans, chronic wasting disease in deer, and bovine spongiform encephalopathy (also known as “mad cow disease”). These diseases are caused by the accumulation of misfolded prion protein (PrP) in the brain. PrP can exist in two distinct conformational states: PrP C , which is the properly folded cellular version of the protein, and PrP Sc , which is the misfolded and infectious conformational isoform. Prions are self-propagating: PrP Sc acts as a template to direct the misfolding of PrP C into additional copies of PrP Sc, resulting in prion amplification and spread throughout the brain.

Although prions are notorious for being infectious (as in the case of variant CJD, which results from the consumption of beef contaminated with mad cow disease prions), the vast majority of CJD cases in humans are sporadic in nature, with no known genetic or environmental cause. Despite decades of intense research efforts, significant questions remain unanswered in prion biology:
- What is the mechanism of spontaneous prion formation in patients with sporadic CJD?
- Why are prions toxic to neurons in the brain?
- Are there any co-factors necessary for prion replication?
My lab uses a combination of mouse, cellular, and in vitro models to study these issues. In particular, we are interested in exploiting the unique properties of the prion protein from the bank vole (Myodes glareolus) in order to gain insight into the biology of prion disease. Bank vole PrP appears to be much more prone to adopting misfolded conformations than PrPs from other species and we have established that transgenic mice expressing bank vole PrP develop a spontaneous neurodegenerative disease that accurately recapitulates most aspects of human prion disease. We are currently using these mice to study the factors that govern spontaneous prion formation in the brain.
Exploring the “Expanding Universe” of Prion Diseases
There is increasing evidence that the aggregation-prone proteins that cause much more common neurodegenerative diseases, such as AD and PD, exhibit certain properties that are reminiscent of prions. For instance, injection of transgenic mice with aggregated a-synuclein protein, which causes PD and related diseases such as Dementia with Lewy bodies and multiple system atrophy, accelerates disease and induces the deposition of a-synuclein in the brain. Similarly, inoculation of transgenic mice with aggregated beta-amyloid (Aβ) peptide, which is one of the two major players in AD, induces the formation of amyloid plaques similar to those found in the brains of AD patients. These results demonstrate that a-synuclein and Abeta aggregates, like prions composed of PrP, are self-propagating.
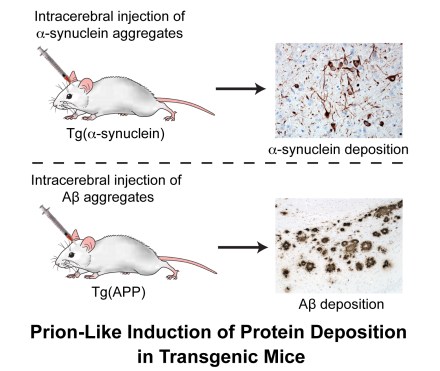
Although it is unlikely that AD and PD are ‘infectious’ diseases in the same sense as the classical prion diseases, there is much to be gained from interrogating the prion-like nature of these illnesses. My lab is interesting in exploring the following questions:
- Are different ‘strains’ of Aβ or alpha-synuclein aggregates responsible for the diverse clinical presentations of disease in humans?
- How do Aβ and alpha-synuclein aggregates spread throughout the brain?
- Can the prion-like properties of alpha-synuclein and Aβ be exploited to enable an earlier diagnosis of disease?
Techniques for Monitoring Neurodegeneration and Protein Misfolding in Living Animals
Although currently available mouse models of AD and PD constitute a powerful tool for studying these diseases, they suffer from some important drawbacks. In particular, it is very difficult to assess the disease status of the mice without resorting to postmortem analysis of brain tissue. Thus, we are interested in developing methods for visualizing protein aggregation and neurodegeneration in the brains of living animals. We have recently demonstrated that transgenic mice expressing a firefly luciferase reporter gene under the control of the glial fibrillary acidic protein (GFAP) promoter permit the kinetics of Aβ deposition in the brain to be monitored in vivo using bioluminescence imaging. My lab is interesting in designing improvements to this paradigm as well as developing novel genetic- and small molecule-based techniques for more directly monitoring protein aggregation in the brain.
