Investigating the Mechanisms of Spontaneous Prion Formation and Prion Neurotoxicity
Prions are infectious proteins that cause a variety of rare but invariably fatal neurodegenerative illnesses such as CJD in humans, chronic wasting disease in deer, and bovine spongiform encephalopathy (also known as “mad cow disease”). These diseases are caused by the accumulation of misfolded prion protein (PrP) in the brain. PrP can exist in two distinct conformational states: PrPC, which is the properly folded cellular version of the protein, and PrPSc, which is the misfolded and infectious conformational isoform. Prions are self-propagating: PrPSc acts as a template to direct the misfolding of PrPC into additional copies of PrPSc, resulting in prion amplification and spread throughout the brain.
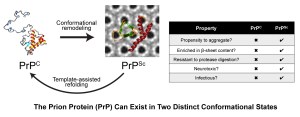
Although prions are notorious for being infectious (as in the case of variant CJD, which results from the consumption of beef contaminated with mad cow disease prions), the vast majority of CJD cases in humans are sporadic in nature, with no known genetic or environmental cause. Despite decades of intense research efforts, significant questions remain unanswered in prion biology:
- What is the mechanism of spontaneous prion formation in patients with sporadic CJD?
- Why are prions toxic to neurons in the brain?
- Are there any co-factors necessary for prion replication?
My lab uses a combination of mouse, cellular, and in vitro models to study these issues. In particular, we are interested in exploiting the unique properties of the prion protein from the bank vole (Myodes glareolus) in order to gain insight into the biology of prion disease. Pioneering work from the labs of Umberto Agrimi and Romolo Nonno [Nonno 2006; Agrimi 2008; Di Bari 2013] have demonstrated that bank voles are highly susceptible to prions from a diverse range of species, suggesting that they do not impose a “species barrier” during prion replication. We recently demonstrated that transgenic mice engineered to express bank vole PrP (BVPrP) in their brains are also highly susceptible to many different prion strains, suggesting that BVPrP may be a “universal acceptor” for prions [Watts 2014]. Unexpectedly, we found that transgenic mice expressing the I109 allotype of BVPrP develop a spontaneous, rapidly transmissible neurodegenerative disease that recapitulates all the neuropathological hallmarks of authentic prion disease, arguing that prions are formed spontaneously in the brains of these mice [Watts 2012].
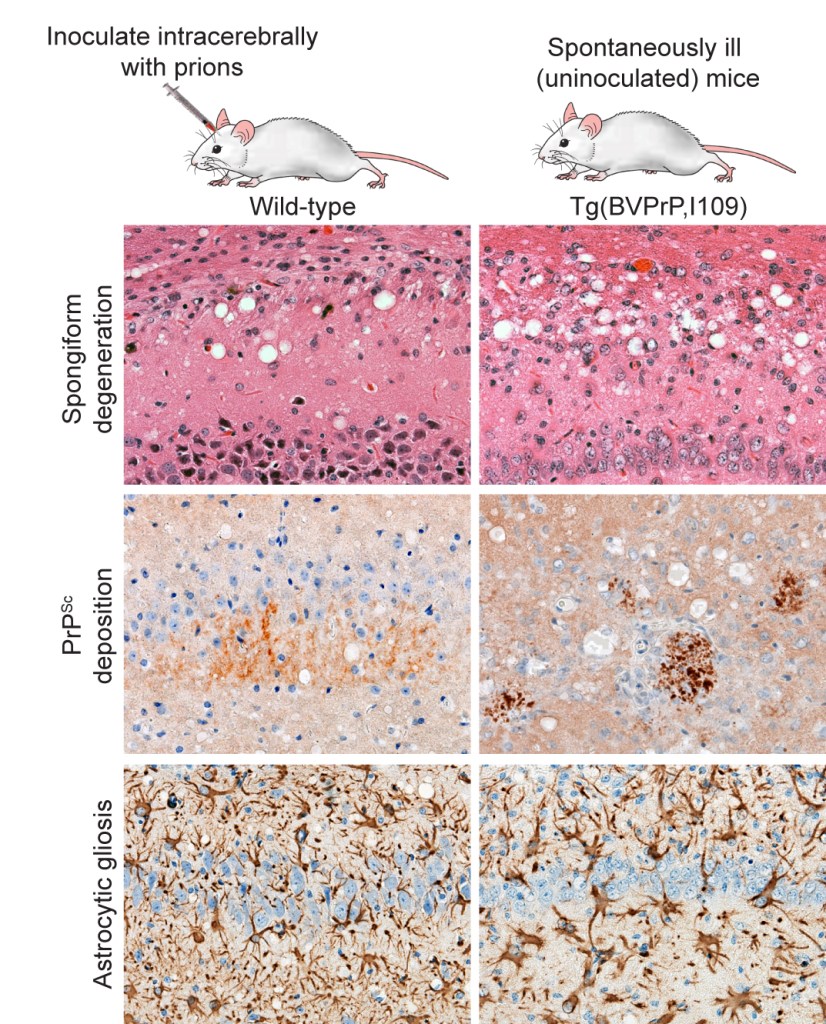
Thus, BVPrP appears to be much more prone to adopting misfolded conformations than PrPs from other species. We are currently using the BVPrP-expressing transgenic mice to study the factors that govern spontaneous prion formation in the brain and to isolate candidate neurotoxic and self-propagating PrP assemblies, which may ultimately lead to new therapeutic strategies for halting the spread of prions in the brain.
